Cystic Fibrosis
What is a Cystic Fibrosis?
Cystic Fibrosis (CF) is a genetic disorder that affects the lungs, pancreas, liver, intestines, and other organs. It is caused by a defective gene that makes the body produce abnormally thick and sticky mucus.
Because CF affects the airways and lungs, which can make breathing difficult and lead to frequent infections, many people consider it to be a lung illness.
However, because it also results in pancreatic cysts and scarring (fibrosis), it is also known as cystic fibrosis. It may be difficult to obtain nutrients through your digestive tract because of this damage and the thick mucus that can obstruct ducts that release digestion enzymes. Your liver, sinuses, intestines, and sex organs may also be impacted by cystic fibrosis.
You have thin, watery mucus lining your organs and cavities, such your nose and lungs. A genetic mutation causes low quantities of specific proteins or proteins that don’t function correctly in patients with cystic fibrosis. These defective proteins cause minerals that thin your mucus by transferring water into it to become trapped inside cells, making the mucus thick and sticky.
Cystic fibrosis is a congenital condition. It’s a chronic condition that worsens with time. The majority of CF patients do not live as long as those without the condition.
The Pathological Process
Cystic Fibrosis is a hereditary condition that mostly affects the lungs, liver, pancreas, and intestines.
It affects the exocrine glands, which produce sweat and mucus. It results in inflammation, lung tissue damage, and an increased risk of bacterial infections.
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) is an aberrant gene that causes the creation of thick, sticky mucus that obstructs the lungs’ airways and frequently causes lung infections. Additionally, the thick mucus obstructs the pancreatic ducts, which prevents the digesting pancreatic enzymes from entering the small intestine and carrying out their regular activity.
Three theories exist:
The lungs’ nutrient-rich mucus builds up and becomes more viscous due to the absence of chloride migration via the CFTR protein, which allows bacteria to evade the body’s immune system.
The CFTR protein deficiency causes a paradoxical increase in the absorption of salt and chloride. Mucus becomes thick and dehydrated as a result of increased water absorption.
According to this view, dry mucus, pancreatic secretion, biliary secretion, and other conditions are caused by abnormal chloride move out of the cell.
According to these beliefs, the main cause of harm in CF patients is the thick secretions that obstruct the affected organs’ narrow channels. Because of the buildup of enzymes and obstructions of the channels, infections result from blockages in the lung airways.
Worldwide Prevalence or Incidence
One case every 3200 white people, one case per 15,000 black people, one case per 9,200 Hispanic people, and one case per 31,00 Asian Americans are the prevalence rates in the United States. Sharma and Girish (2008).
According to another study, 70,000 people worldwide and about 30,000 children and adults in the US have cystic fibrosis (CF) (Grief, 2008). Although CF is lethal, the survival rate has gone up due to advancements in medical research and treatment methods. Over 40% of people with CF are at least 18 years old.
Cystic fibrosis types
Cystic fibrosis comes in two varieties:
Multiple organs are frequently affected by classic cystic fibrosis. Usually, a diagnosis is made during the first few years of life.
A less severe version of the illness is called atypical cystic fibrosis. One organ may be the sole one affected, or symptoms may appear and disappear. Usually, older kids or adults are diagnosed with it.
Cystic fibrosis (CF) symptoms
Symptoms of cystic fibrosis include:
- chronic lung conditions such pneumonia or bronchitis.
- Loose or greasy stool.
- Breathing difficulties.
- Frequent wheezing.
- Frequent infections of the sinuses.
- A persistent cough.
- Slow growth.
- Failure to thrive, or the failure to put on weight even when eating a healthy diet and getting enough calories.
Atypical symptoms of cystic fibrosis:
Some of the symptoms of traditional cystic fibrosis may also be present in people with atypical CF. Over time, you may also encounter:
- Persistent sinusitis.
- Polyps in the nose.
- Heatstroke or dehydration caused by incorrect electrolyte levels.
- Diarrhea.
- pancreatitis.
- Unintentional weight loss.
Cystic fibrosis: what causes it?
Cystic fibrosis is caused by variations or mutations in the CFTR gene. On the cell surface, CFTR produces a protein that functions as an ion channel. Certain chemicals can flow through ion channels, which function similarly to gates in a cell’s membrane.
For chloride ions, a class of mineral with a negative electrical charge, CFTR typically creates a gate. Mucus is thinned and made more slick by chloride, which leaves the cell with water. This is prevented in CF patients by CFTR gene mutations, which keeps the mucus thick and sticky.
CFTR gene mutations are classified into classes I through VI based on their respective effects. others create proteins that don’t function correctly, others make very little protein, and some produce no protein at all.
The most recent classification scheme groups mutations according to the issues they cause with the CFTR protein’s production:
- Class 1 Mutations in the synthesis of proteins
- Class 2 Mutations in protein processing
- Class 3 gating mutations
- Class 4 conduction mutations
- Class 5 Not enough mutations in proteins
Class 1 Mutations in Protein Production
Mutations affecting protein synthesis, such as splice and nonsense mutations, prevent the CFTR protein from being produced.
Amino acids, the building blocks of all proteins, including CFTR, are joined to form a lengthy chain. The cell knows which of the 20 available amino acids to employ at each location in the chain thanks to the protein-building instructions encoded in the CFTR gene. The gene’s letters also form a “stop” signal, which informs the cell that it has finished the instructions and can cease producing the protein.
The protein-building instructions include an early stop signal that results in the premature termination of CFTR protein production in the event of a nonsense mutation in the CFTR gene. As a result, the cell starts producing the CFTR protein as usual up to the early stop signal.
The cell prematurely halts synthesis because it “thinks” that the instructions have been completed. CFTR does not produce a functional protein because the cell stops reading the instructions before it completes the protein synthesis.
The signal that tells the cell where the unnecessary letters in the instructions start or stop is altered by a splice mutation. The cell can no longer determine where to start and stop reading when it attempts to read its RNA copy of the instructions.
The cell will consequently either eliminate some pertinent letters or leave in some irrelevant ones. The cell will be unable to produce the proper CFTR protein if it attempts to follow the RNA instructions that either contain irrelevant letters or omit pertinent ones.
In a healthy individual, segments of DNA letters that do not code for protein interrupt the instructions written in a gene, much way advertisements may interrupt a magazine article. A unique signal is used to indicate the start and finish of these unnecessary letter segments.
The cell converts the DNA letters into a similar alphabet called ribonucleic acid (RNA) in order to produce the protein. It then follows the signals to remove all of the unnecessary letters, just like you might remove advertisements. In this manner, it is possible to read the instructions from beginning to end.
Class 2 Mutations in Protein Processing
The 1,480 amino acids that make up the CFTR protein. The CFTR protein takes on a stable three-dimensional form when it is assembled with all of the appropriate amino acids. For chloride to be transported, it must have the proper form.
The CFTR protein is unable to form its proper 3-D shape and function when a mutation results in the deletion of an amino acid or the addition of an erroneous amino acid. These mutations are classified as mutations that affect how proteins are processed.
F508del, the most prevalent CF mutation, is typically regarded as a processing mutation. The F508del mutation causes the CFTR protein to lose one amino acid. The CFTR protein cannot maintain its proper three-dimensional form without this building ingredient. The cell gets rid of the protein after realizing it isn’t in the proper structure.
By allowing the CFTR protein with the F508del mutation to fold in a more proper form, the medication combination activates the protein, allowing more chloride to pass through. This medicine combination aids in the movement of some chloride by the mutant CFTR protein, although it is not a perfect solution. This chloride migration lessens CF symptoms.
Missense mutations, in addition to F508del, can occasionally result in processing issues and, in those situations, may be regarded as processing mutations. When an erroneous amino acid is integrated into the CFTR protein due to a change in DNA letters, this is known as a missense mutation. This results in either a decrease in the protein’s function (defective gating or conduction) or its abundance at the cell surface (defective processing).
Class 3 Mutations in Gating
The structure of the CFTR protein resembles a tunnel or channel with a gate. When chloride must pass through the channel, the cell can open the gate. The gate remains closed otherwise.
Gating mutations prevent chloride from passing through by locking the gate in the closed position. By keeping the CFTR channel’s gate open, the medication aids patients with gating mutations. This lessens CF symptoms by allowing chloride to pass through the channel.
Class 4 Mutations in Conduction
A mutation in one of the CFTR’s amino acids can occasionally cause the protein to function less properly even when it forms the proper 3-D structure. Chloride must be able to pass through the protein’s channel swiftly and smoothly for CFTR to function properly. Certain mutations alter the channel’s internal geometry, making it more difficult for chloride to pass through. We refer to this type of mutation as a conduction mutation.
Class 5 Not Enough Protein Mutations
There is less of the typical CFTR protein on the cell surface as a result of insufficient protein mutations. A tiny quantity of CFTR protein is created, just a small portion of the cell surface protein functions properly, or normal cell surface protein breaks down too quickly, leaving only a small amount of protein left. These are some of the possible causes of this.
In each instance, the chloride channel only partially or partially functions due to a lack of functional proteins on the cell surface. Missense and splice mutations are two of the many mutations that might result in insufficient protein.
Issues related to CF?
Among the complications of CF are:
- Infections: Bacteria that you are unable to expel from your lungs and airways can be trapped there by thick mucus. Frequent infections may result from this.
- Congenital bilateral vas deferens absence (CBAVD): Males with this disorder lack the vas deferens, or sperm ducts. If they wish to have biological children, they frequently require the assistance of reproductive treatments.
- Diabetes: Diabetes associated with cystic fibrosis can be caused by damage to the pancreas.
- Malnourishment: You may be at risk for malnutrition if you have thick mucus in the intestines and insufficient pancreatic enzymes to aid in digestion.
- Both osteoporosis and osteopenia: Conditions that cause your bones to become too thin can result from your digestive tract’s incapacity to absorb nutrients.
- Complications throughout pregnancy: Inadequate nutrition can result from CF’s effects on the digestive system. The chance of pregnancy difficulties may rise as a result. The most frequent complication is preterm birth, or early birth.
Testing and Diagnosis
How is a diagnosis of cystic fibrosis made?
Cystic fibrosis is frequently tested for by medical professionals as part of a newborn screening. A few drops of blood taken from your baby’s heel are used by the providers to conduct this test. Your pancreas produces a substance called immuno-reactive trypsinogen (IRT), which a lab tests for in a blood sample. IRT levels in the blood are greater in CF patients. IRT testing is frequently performed on newborns within a few weeks of their birth.
IRT levels can be elevated by certain situations, such as preterm birth. A positive IRT test does not, therefore, indicate that your child has cystic fibrosis. Your doctor will order more tests to reach a final diagnosis if your baby’s IRT levels are greater than anticipated.
The newborn test fails to identify high IRT levels in a person with cystic fibrosis in approximately 5% of instances. It’s also possible that you were born before regular CF screening became accessible. A healthcare professional will conduct a sweat test and, if necessary, additional tests if you or your child exhibit symptoms of cystic fibrosis.
Cystic fibrosis tests
- Sweat test: The sweat test calculates how much chloride is present in your perspiration. Those with cystic fibrosis have greater sweat chloride levels. Although it is the most definitive test for CF, individuals with atypical CF may have benign results.
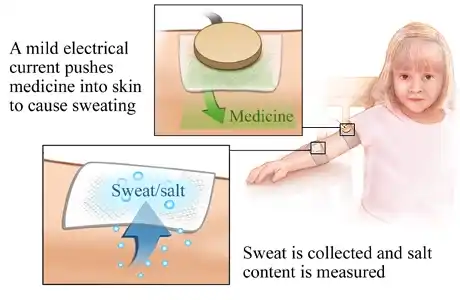
- Genetic examinations: Blood samples are tested for alterations in the genes that cause cystic fibrosis.
- Imaging tests, such as chest and sinus X-rays, are used by providers to validate or support a diagnosis of cystic fibrosis. CF cannot be diagnosed by imaging alone.
- Testing for pulmonary function: These tests assess the function of your lungs.
- Sputum culture: Sputum, or mucus that has been coughed up from your lungs, is sampled by your healthcare professional, who then examines it for microorganisms. Pseudomonas is one of the bacteria that is most frequently discovered in CF patients.
- A pancreatic biopsy: This can let your doctor know if you have pancreatic damage or cysts.
- Differential in nasal potential (NPD): The little electrical charge that is often found in the lining of your nose is measured by this test. This charge is produced by ion mobility. Because of the way CF affects their ion channels, people with CF have less ion mobility.
- Measurement of intestinal current (ICM): To conduct this test, a healthcare professional obtains a sample of rectal tissue. The amount of chloride secreted by the sample is measured by a laboratory.
Treatment of Cystic Fibrosis
How is cystic fibrosis treated?
A cure for cystic fibrosis does not exist. A cystic fibrosis specialist and other healthcare professionals can help you manage the illness and its symptoms. Management entails:
- Using breathing exercises and tools to break up mucus will help you keep your airways open and clear.
- Drugs known as CFTR modulators that aid in resolving problems with CFTR proteins.
- Drugs that lessen particular symptoms.
- Ensuring that you eat enough of the appropriate kind of calories.
- Surgery.
Techniques for clearing the airways
If you have cystic fibrosis, there are several methods you may assist maintain clear airways:
Techniques for breathing and coughing.
You can learn ways to clear your airways and reduce mucus from a physical therapist who specializes in treating CF.
PEP stands for positive expiratory pressure: PEP devices are worn in your mouth or on your face as a mask. By keeping your airways open and pushing mucus out, they create resistance that makes it harder to exhale. Certain PEPs, such as oscillating PEP devices (Flutter), vibrate to break up mucus.

Vests for airway clearance: An inflated vest that fastens to a machine is known as an airway clearance vest or high-frequency chest wall oscillation device. To break up mucous, the vest vibrates.
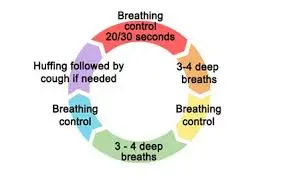
The Active Cycle of Breathing Techniques (ACBT) is a set of breathing techniques that include thoracic expansion, forceful expiration (huffing), and breathing regulation in order to mobilize and remove secretions.
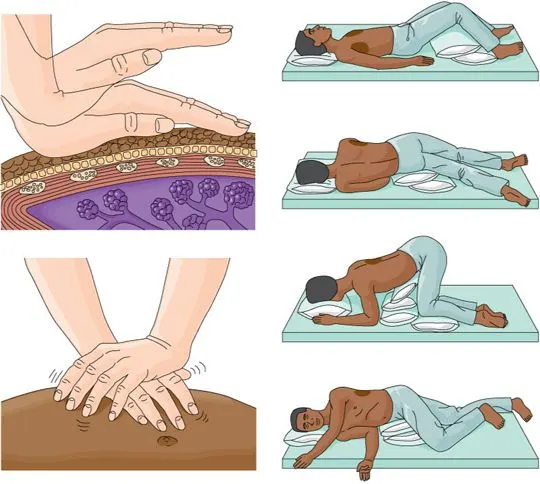
Drainage of Posture
Postural drainage, which involves positioning the body so that secretions can gravitationally flow from the smaller airways into the major airway, is a crucial component of cystic fibrosis treatment. Which particular postural drainage positions are required will be taught and advised by the physical therapist.
Vibration and Percussion
These methods are employed to mobilize and loosen secretions. The physical therapist can suggest sitting, lying on either side, or on your front or back.
The technique known as “chest clapping,” or percussion, involves rhythmically and firmly clapping the chest over a layer of clothing or a towel for 15 to 20 seconds, pausing for at least five seconds, over the area that needs to be drained.
At the conclusion of the position, vibrations or “chest shaking” are performed. As the patient exhales, it describes brief, rhythmic squeezes on the chest wall.
Inhibitors of CFTR for cystic fibrosis
Drugs known as CFTR modulators can help fix problems with proteins produced by mutant CFTR genes and boost the quantity of functional proteins on the surface of your cells. They don’t treat cystic fibrosis. However, some people have had significant improvements in their life expectancy and symptoms.
Nevertheless, some CF patients are ineligible for or unable to tolerate modulator treatments.
Among the CFTR modulators are:
- Ivacaftor
- Ivacaftor/lumacaftor,
- (ivacaftor/tezacaftor)
- (ivacaftor/tezacaftor/elexacaftor)
Additional drugs for people with cystic fibrosis
Additionally, your doctor can recommend drugs to control symptoms, treat infections, or lessen inflammation. These consist of:
Antibiotics. Antibiotics may be prescribed by your doctor for the prevention or treatment of infections.
Bronchodilators inhaled. Through the opening and relaxation of your airways, bronchodilators facilitate breathing.
Hypertonic saline was inhaled. Saline solutions’ salt draws water, and which thins mucus and facilitates its removal.
Medications that reduce inflammation. These drugs lessen edema. These consist of nonsteroidal anti-inflammatory medications (NSAIDs) and corticosteroids.
Enzymes found in the pancreas. These aid in food digestion and nutrient absorption.
stool softeners. These can ease constipation and facilitate bowel movements.
The diet for cystic fibrosis
Your dietary requirements differ from those of a person without cystic fibrosis (CF). Enzymes that aid in food digestion may not be produced or secreted by your pancreas if you have cystic fibrosis.
This indicates that nutrients and fats from meals are not completely absorbed by your intestines.
A certified dietician or your CF specialist could suggest a nutrition plan. It might consist of:
Consuming more calories every day. Compared to someone without CF, this could be up to twice as many calories.
Consuming foods heavy in fat. This is crucial for increasing your intake of fat-soluble vitamins.
Keeping a weight that is higher than normal from an early age. Growing taller and having bigger lungs can assist alleviate symptoms as you get older.
Taking pills of an enzyme supplement. Supplemental enzymes aid in meal digestion.
consuming more salt. This aids in replenishing the extra salt lost through perspiration. This is particularly crucial when exercising and in hot, muggy weather. Find out from your provider how much salt you require on a daily basis.
Cystic fibrosis surgeries
If you have cystic fibrosis or one of its consequences, you might require surgery. These could consist of:
- Either sinus or nasal surgery.
- Surgery to eliminate obstructions in the bowel.
- Lung replacement.
- Transplantation of the liver
Prevention
Is CF preventable?
It cannot be prevented because you are born with cystic fibrosis. Ask your healthcare practitioner regarding prenatal genetic testing or the likelihood that your biological children will have cystic fibrosis (CF) if you carry a variation of the CFTR gene.
Prognosis
The prognosis for cystic fibrosis has improved as a result of improved access to healthcare, better treatment, and earlier detection through screening. In the United States, children with cystic fibrosis had a median survival age of six months in 1959.
The predicted survival rates for men and women in 2010 were 40 and 37 years, respectively. The median lifespan in Canada rose from 24 years in 1982 to 47.7 years in 2007. When treated at specialty clinics, people with CF born in the United States in 2016 are expected to live for 47.7 years.
In recent years, life expectancy has quickly increased due to the introduction of novel medications, such as CFTR modulators.
Given the low number of deaths among people with cystic fibrosis each year, projections of the median expected life expectancy in 2020 were imprecise, but it was approximately 59 years.
As of 2009, 92% of CF patients over the age of 18 in the US had completed high school, 67% had at least some college education, 15% had a disability, 9% were unemployed, 56% were single, and 39% were married or cohabitating.
Quality of life
Managing chronic conditions can be challenging. CF is a long-term disease that impacts the respiratory and digestive systems, leading to persistent respiratory infections and widespread malnourishment.
The lungs’ airways get blocked by the thick secretions, which frequently results in inflammation and serious lung infections. When it is affected, it impacts a person with cystic fibrosis’s quality of life and their capacity to perform daily duties.
CF patients can improve their quality of life in a variety of ways. The goal of exercise is to improve lung function. A CF patient’s quality of life can be greatly enhanced by including an exercise program into their daily routine.
Although there is currently no known cure for cystic fibrosis (CF), a variety of drugs are used to treat the condition. These include steroids, bronchodilators, mucolytics, and antibiotics. These drugs are used to enlarge airways, reduce inflammation, fight lung infections, and loosen mucus, respectively.
FAQs
What is cystic fibrosis primary cause?
Mutations in a gene known as the cystic fibrosis transmembrane conductance regulator (CFTR) produce the hereditary disease known as cystic fibrosis. The CFTR protein is instructed by the CFTR gene. A person will have cystic fibrosis if they inherit two copies of a mutant CFTR gene, one from each of their biological parents.
In what way is cystic fibrosis diagnosed?
A sweat test looks for elevated chloride levels in your perspiration. The most common test for identifying cystic fibrosis is the sweat test. It can be used to confirm a positive diagnosis from a newborn screening or if you or your kid exhibit symptoms that might point to cystic fibrosis.
What are CF early warning indicators?
Fever, possibly accompanied by sweating at night. digestive problems such constipation, persistent (long-term) diarrhea, or excruciating stomach discomfort. lung and sinus infections. Yellowing of the skin and eyes for an unusually extended period of time after delivery is known as jaundice.
Which bodily parts are impacted by cystic fibrosis?
Thick mucus produced by cystic fibrosis clogs the intestines, pancreas, and lungs. Malnutrition, stunted growth, recurrent respiratory infections, breathing difficulties, and chronic lung disease might result from this. Every state in the US mandates CF testing for infants.
Is hair loss a symptom of cystic fibrosis?
Changes in our bodies caused by CF and the drugs it demands are among the issues that many of us with the condition face. Common problems that can arise include bruises, acne outbreaks, hair loss, water retention, and bloating.
When does CF begin to manifest?
The majority of children are now diagnosed with cystic fibrosis (CF) within the first month of their lives, frequently before they exhibit any symptoms, thanks to a rise in newborn screening programs. Children with cystic fibrosis are often diagnosed by the age of two.
Is there a birth CF test?
At five days of age, newborns are offered newborn screening, often called the heel prick test, to check for a number of rare but dangerous illnesses, including cystic fibrosis (CF). A positive newborn screening result may indicate that a baby has cystic fibrosis (CF), but follow-up testing may not confirm a diagnosis.
Reference
- Types of CFTR mutations. (n.d.). Cystic Fibrosis Foundation. https://www.cff.org/research-clinical-trials/types-cftr-mutations
- Cystic fibrosis. (2025, February 7). Cleveland Clinic. https://my.clevelandclinic.org/health/diseases/9358-cystic-fibrosis
- Seo, C. (2023, January 13). Physical Therapy management of cystic fibrosis. Scandinavian Physical Therapy Center, Dubai. https://scandinavianphysical therapy center.com/blog/physical therapy -management-of-cystic-fibrosis



One Comment